ArriVent Biopharma IPO investment analysis
January 8, 2024
This is not investment advice. We used AI and automated software tools for most of this research. A human formatted the charts based on data / analysis from the software, prompted the AI to do some editing, and did some light manual editing. We did some fact checking but cannot guarantee the accuracy of everything in the article. We do not have a position in or a relationship with the company.
Overview
Arrivent BioPharma is a clinical-stage biopharmaceutical company developing innovative therapies for cancer. Furmonertinib, Arrivent's lead candidate, is an EGFR tyrosine kinase inhibitor (TKI) showing selectivity towards mutated EGFR forms in non-small cell lung cancer (NSCLC). It aims to target a broader spectrum of EGFR mutations beyond those addressed by existing EGFR TKIs.
Currently, furmonertinib is involved in several clinical trials, including a pivotal Phase 3 trial (FURVENT) evaluating it as a first-line therapy for NSCLC with specific mutations like the EGFR exon 20 insertion.
Furmonertinib is approved in China by Shanghai Allist Pharmaceuticals Co. Ltd. for classical EGFR mutant NSCLC. However, it is not yet approved elsewhere. Arrivent holds the rights to furmonertinib's development and commercialization worldwide, excepting greater China, a license obtained from Allist in 2021.
Preliminary data, particularly from the Chinese FAVOUR trial, indicates promising activity for furmonertinib, with significant tumor reduction observed in patients. The drug has shown potential efficacy for patients with uncommon EGFR mutations such as exon 20 insertions or PACC mutations, inadequately addressed by current treatments. While interim data is promising, it may not necessarily reflect final trial outcomes, which could affect development strategies and commercial plans for the drug.
Pipeline overview
| Product name | Modality | Target | Indication | Discovery | Preclinical | Phase 1 | Phase 2 | Phase 3 | FDA submission | Commercial |
|---|---|---|---|---|---|---|---|---|---|---|
| Furmonertinib | Small molecules | EGFR Inhibitor | First-line NSCLC with EGFR Exon 20 Insertion Mutations | |||||||
| Furmonertinib | Small molecules | EGFR Inhibitor | First-line or greater NSCLC with EGFR PACC mutations | |||||||
| Furmonertinib | Small molecules | EGFR Inhibitor | Second-line or greater NSCLC classical EGFR mutations | |||||||
| ARR-002 | Antibody-drug conjugate | Undisclosed | Oncology |
Highlights and risks
EGFR is a well-validated target in NSCLC
Clinical data thus far suggests drug has anti-tumor activity and activity against target mutations
Better activity against brain metastases is a potential differentiator
Potential use in second-line classical EGFR mutations provides opportunity for market expansion
EGFR TKIs are an established and highly competitive drug class
Drug will require competitive clinical data to expand clinical use beyond low-frequency mutations or later-line therapy
Initial indications are targeting relatively small patient populations
Valuation
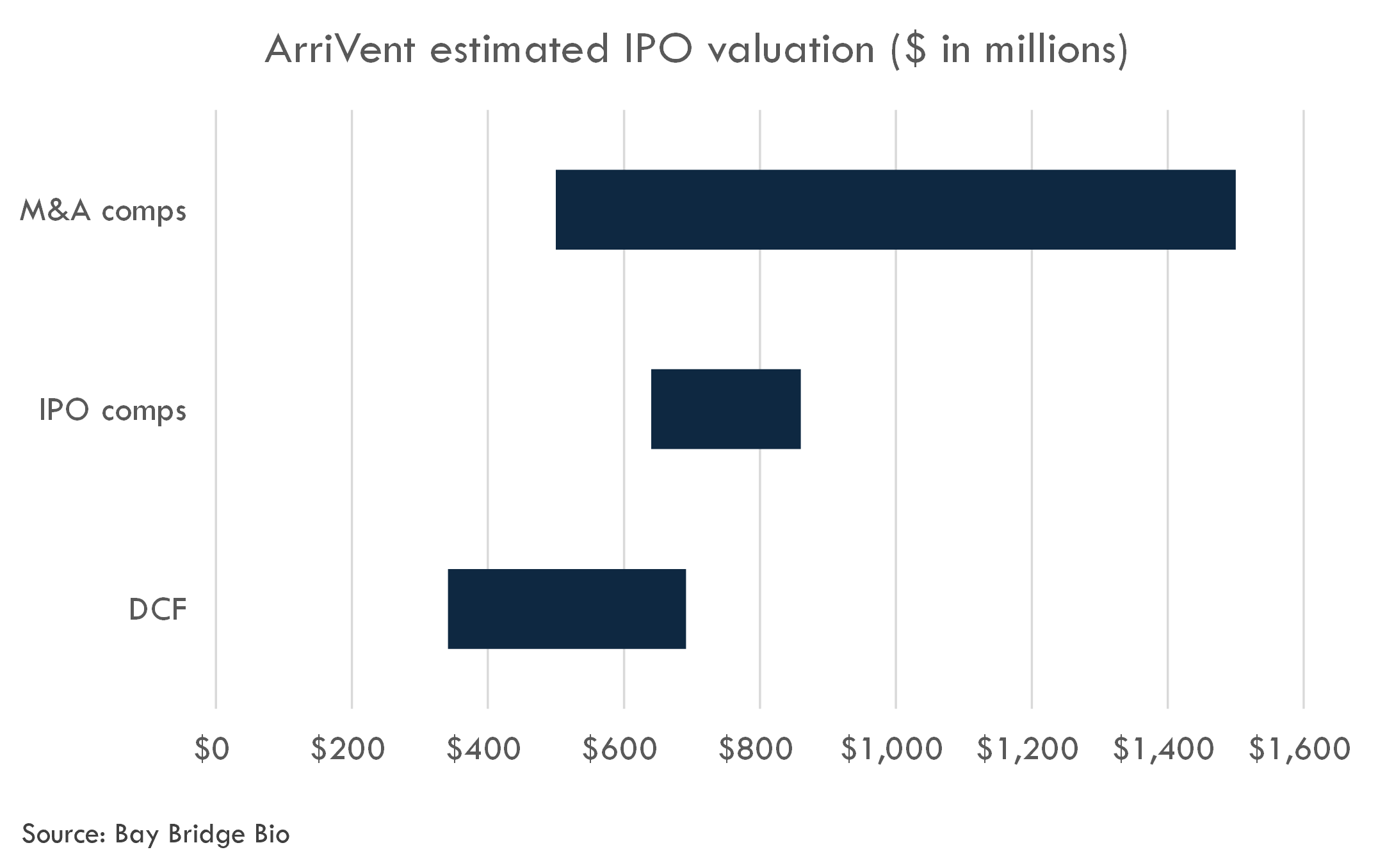
ArriVent filed an S-1 in January 2024. We estimate the valuation of their last round at $400 million. Based on comparable IPOs, we estimate an IPO fully-diluted post-money equity value of $640-860 million.
Potential investors in ArriVent's IPO include:

Weekly analyses of biotech startups, generated by AI
Receive high quality, AI-generated analyses of biotech startups, public companies and scientific papers each week.
Furmonertinib
Scientific thesis
The therapeutic rationale for using an EGFR tyrosine kinase inhibitor (TKI) such as furmonertinib in various settings of non-small cell lung cancer (NSCLC) with specific EGFR mutations is grounded in the biology of how these mutations drive cancer growth and how targeted therapies can inhibit these pathways.
First-line NSCLC with EGFR Exon 20 Insertion Mutations: EGFR exon 20 insertion mutations result in a change in the structure of the EGFR kinase domain, leading to increased activity and driving oncogenesis. Standard EGFR TKIs are often less effective against exon 20 insertions because of the steric hindrance within the ATP binding pocket of the mutant EGFR. Furmonertinib, with its irreversible binding to the EGFR kinase domain and its ability to penetrate the blood-brain barrier, provides a strong rationale for use as a first-line agent in this subset of patients. Preliminary clinical data suggesting tumor size reduction in patients and the FDA Breakthrough Therapy Designation underscore its potential effectiveness in this setting.
First-line or greater NSCLC with EGFR PACC mutations: EGFR PACC mutations are uncommon EGFR mutations that may not be effectively targeted by currently approved EGFR TKIs. Since furmonertinib demonstrated preclinical activity against these mutations, there is a rationale to investigate its use in patients harboring EGFR PACC mutations. The ongoing FURTHER Phase 1b trial aims to establish the proof of concept for the use of furmonertinib in this patient population.
Second-line or greater NSCLC with classical EGFR mutations: Classical EGFR mutations (such as deletions in exon 19 and the L858R point mutation in exon 21) lead to constitutive activation of EGFR signaling pathways, driving cell proliferation and survival in NSCLC. Furmonertinib is already approved in China for first-line treatment in this group of patients, providing an argument for its clinical utility. Its efficacy in second-line or greater settings is of interest for those who may develop resistance to first-line therapies, including the common T790M resistance mutation.
The rationale behind the use of furmonertinib involves several important factors:
- Its selective inhibition of mutant EGFR, potentially reducing off-target effects and toxicity while effectively targeting cancer cells.
- Its capacity to produce sustained target inhibition by forming irreversible bonds with the EGFR kinase domain.
- Its ability to penetrate the central nervous system (CNS), which may be critical for treating patients with brain metastases.
- Its observed preclinical and clinical activity against specific uncommon EGFR mutations that are not well served by currently available medications.
Future clinical trials like the FURVENT Phase 3 trial and FURTHER Phase 1b will provide more definitive data on the effectiveness and safety profile of furmonertinib in the respective mutation-specific patient populations, potentially leading to wider use and approvals in other parts of the world beyond China.
Background on EGFR TKIs
The science underlying the use of EGFR tyrosine kinase inhibitors (TKIs) in NSCLC is well-established, with multiple EGFR inhibitors approved for clinical use. However, the effectiveness of these inhibitors can vary significantly depending on the specific EGFR mutations present in the tumor.
Established Science:
-
EGFR Function and Mutation-Driven Oncogenesis: The role of EGFR as an oncogene in NSCLC and the way EGFR mutations lead to activation of downstream signaling pathways is well-understood. Classical EGFR mutations, like exon 19 deletions and L858R, are well established as drivers of NSCLC and predictive biomarkers for the efficacy of EGFR TKIs.
-
EGFR TKIs in NSCLC: The therapeutic benefit of EGFR TKIs for patients with classical EGFR mutations is a standard of care in first-line metastatic NSCLC and is supported by a wealth of evidence and numerous clinical trials.
-
Blood-Brain Barrier Penetration and CNS Activity: EGFR TKIs like osimertinib have been shown to penetrate the blood-brain barrier and are effective against CNS metastases. This is one of the rationales for the development of furmonertinib and its expected similar CNS penetration.
-
Resistance Mechanisms: It's broadly accepted that cancer cells can develop resistance to EGFR TKIs, such as by acquiring secondary mutations like T790M, which led to the development of third-generation EGFR TKIs, targeting these resistant mutations.
Uncertainty and Scientific Debate:
-
Uncommon Mutations: The effectiveness of EGFR TKIs for less common mutations, like exon 20 insertions and EGFR PACC mutations, is less established. Although preclinical studies and early clinical trials (such as the FAVOUR trial for furmonertinib) indicate potential efficacy, these findings are still subject to confirmation in larger, rigorous clinical trials.
-
Optimal Dosing and Toxicity: The best dose of furmonertinib to maximize its efficacy while minimizing side effects, particularly for different mutation subsets, remains to be fully determined. Data from ongoing trials are expected to shed light on this.
-
Differentiating Between TKIs: While furmonertinib is designed to be effective across a broader range of EGFR mutations, direct comparisons with other EGFR TKIs in terms of efficacy and safety are still necessary to confirm its position in the treatment landscape.
-
Brain Metastasis Responses: Even though preclinical models show furmonertinib's ability to cross the blood-brain barrier, actual clinical efficacy in humans, especially in comparison with other EGFR TKIs like osimertinib, requires further clinical data.
The epidermal growth factor receptor (EGFR) is a well-established therapeutic target in non-small cell lung cancer (NSCLC), particularly for tumors that harbor activating mutations in the EGFR gene. The scientific literature extensively supports the role of EGFR and its mutations in the pathogenesis of NSCLC and the therapeutic efficacy of EGFR tyrosine kinase inhibitors (TKIs). Below are examples of these literature findings:
First-line NSCLC with EGFR Exon 20 Insertion Mutations:
Exon 20 insertions in the EGFR gene represent approximately 4-10% of all EGFR mutations in NSCLC. These mutations lead to the constitutive activation of the EGFR signaling pathway, driving tumorigenesis. However, exon 20 insertions are less responsive to first-generation and second-generation EGFR TKIs due to structural hindrance that prevents drug binding.
Reference:- Yasuda, H., Park, E., Yun, C. H., Sng, N. J., Lucena-Araujo, A. R., Yeo, W. L., ... & Engelman, J. A. (2013). Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Science translational medicine, 5(216), 216ra177. DOI: 10.1126/scitranslmed.3007205.
First-line or greater NSCLC with EGFR PACC mutations:
EGFR P-loop and αC-helix compressing (PACC) mutations are less characterized in the literature and represent a rarer subgroup of EGFR mutations. There is ongoing research to better understand the biology of these mutations and their response to EGFR TKIs. Preclinical studies have shown that certain EGFR TKIs may have activity against some of these uncommon mutations.
Reference:- Arcila, M. E., Nafa, K., Chaft, J. E., Rekhtman, N., Lau, C., Reva, B. A., ... & Ladanyi, M. (2013). EGFR exon 20 insertion mutations in lung adenocarcinoma: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Molecular cancer therapeutics, 12(2), 220-229. DOI: 10.1158/1535-7163.MCT-12-0620
Second-line or greater NSCLC classical EGFR mutations:
Classical EGFR mutations, such as deletions in exon 19 and L858R point mutations in exon 21, have been comprehensively studied in the literature. First-line treatment of patients with NSCLC harboring these mutations with EGFR TKIs has dramatically improved patient outcomes relative to traditional chemotherapy.
However, patients inevitably develop resistance, often due to secondary EGFR mutations like T790M. This has led to the development of later-generation EGFR TKIs that can overcome certain resistance mechanisms.
Reference:- Mok, T. S., Wu, Y. L., Ahn, M. J., Garassino, M. C., Kim, H. R., Ramalingam, S. S., ... & Blackhall, F. (2017). Osimertinib or platinum–pemetrexed in EGFR T790M–positive lung cancer. New England Journal of Medicine, 376(7), 629-640. DOI: 10.1056/NEJMoa1612674.
These references include clinical trial results and molecular characterization studies that provide evidence for the efficacy of EGFR TKIs in specific NSCLC patient populations. Further studies are ongoing to outline the full spectrum of responsiveness of various EGFR mutations to different generations of EGFR TKIs and to enhance the strategies for targeting these mutations in the clinic.
The evidence base supporting the therapeutic rationale for using epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in various forms of NSCLC has several strengths and weaknesses.
Strengths:
-
Solid Clinical Trial Data for Classical Mutations: The therapeutic efficacy of EGFR TKIs in patients with classical EGFR mutations, such as exon 19 deletions and L858R point mutations in exon 21, is supported by numerous high-quality randomized clinical trials, systematic reviews, and meta-analyses. This constitutes a strong evidence base for the first-line use of EGFR TKIs in these populations.
-
Clear Molecular Targets: The molecular mechanisms whereby EGFR mutations lead to NSCLC are well understood. This provides a clear rationale for targeting the EGFR pathway in these cancers with TKIs.
-
Agreement Across Regulatory Bodies: Multiple EGFR TKIs have been approved by the FDA and other regulatory agencies for first-line treatment in patients with classical EGFR mutations, demonstrating a consensus on the strength of the evidence.
-
Continued Innovation: The development of third-generation TKIs has successfully addressed resistance mechanisms such as the T790M mutation, reflecting the dynamic and responsive nature of the field to emerging scientific evidence.
Weaknesses:
-
Less Evidence for Uncommon Mutations: For rarer mutations like the EGFR exon 20 insertions and PACC mutations, the evidence is not as robust or extensive. Data primarily come from smaller studies, case series, or preclinical models, and thus the level of evidence is lower than for more common mutations.
-
Clinical Efficacy and Safety for Newer Inhibitors: For newer inhibitors targeting a broad range of EGFR mutations, such as furmonertinib, clinical data are still emerging. The strength of the clinical evidence is not yet at the level of more established agents due to ongoing evaluation in Phase 3 trials and limited long-term follow-up data.
-
Resistance and Disease Progression: While EGFR TKIs show good initial responses, resistance development remains a significant issue, and the evidence for the best subsequent treatment approaches is evolving.
-
Generalizability of Study Results: Clinical trials conducted in a specific demographic or ethnic group may not be wholly generalizable to all populations. This needs to be addressed through more inclusive research.
-
Biomarker Utilization: While EGFR mutation status is a key biomarker for the use of TKIs, the optimal use of additional biomarkers (e.g., PD-L1 expression, tumor mutational burden) in conjunction with EGFR status to tailor therapy remains a subject of investigation.
-
Central Nervous System (CNS) Efficacy: CNS efficacy, particularly for newer EGFR inhibitors like furmonertinib, is based on preclinical predictions and early clinical data. Detailed and high-quality evidence from large-scale studies is needed to establish such penetration and efficacy solidly.
Overall Level of Evidence:
The overall level of evidence supporting the use of EGFR TKIs for classical mutations in NSCLC is high, backed by randomized controlled trials and meta-analyses. For furmonertinib and other newer agents' effectiveness against uncommon EGFR mutations, preclinical data, and early clinical results are promising but would be considered lower on the evidence hierarchy until supported by more extensive Phase 3 trial data. The process of furmonertinib's development and the observed preclinical and interim clinical results are encouraging, but comprehensive clinical trial outcomes (like those expected from the FURVENT and FURTHER trials) and further regulatory review are needed to fully validate the drug for broader clinical use.
Clinical trial overview
Furmonertinib is currently being studied in several global clinical studies, and has completed a Phase 3 program in China, where the drug is approved.
FURLONG Phase 3 study (completed)
The FURLONG clinical trial was a Phase 3 trial conducted in China that compared the efficacy and safety of furmonertinib to gefitinib as a first-line treatment for patients with locally advanced or metastatic NSCLC with classical EGFR mutations. A total of 358 patients were randomized in a 1:1 ratio to receive either 80 mg of furmonertinib or 250 mg of gefitinib. The study supported the 2022 approval of furmonertinib for this patient population in China.
Efficacy Results:
The median progression-free survival (PFS) for patients treated with furmonertinib was significantly longer at 20.8 months compared to 11.1 months for those receiving gefitinib, with a hazard ratio of 0.443 (p < 0.0001), indicating a reduction in the risk of disease progression or death by 55.7% with furmonertinib.
The objective response rate (ORR) for furmonertinib was higher at 89% compared to 84% in the gefitinib arm.
Furmonertinib showed a particularly strong efficacy in treating CNS metastases, with a 91% ORR in patients with measurable CNS lesions versus 65% for gefitinib. For patients with measurable or non-measurable CNS metastases, the median CNS-specific PFS was 20.8 months with furmonertinib compared to 9.8 months with gefitinib.
None of the patients treated with furmonertinib who did not have CNS metastases at baseline developed new CNS lesions during the trial, whereas 8 patients treated with gefitinib did.
Safety Results:
Furmonertinib had a lower incidence of Grade 3 treatment-related adverse events (TRAEs) at 11.2% compared to 17.9% with gefitinib.
Serious TRAEs were also slightly lower with furmonertinib at 5.6% versus 6.1% for gefitinib.
Notable TRAEs with furmonertinib included increased liver enzymes, cerebral infarction, and gastrointestinal issues.
The rate of trial discontinuation due to TRAEs was low at 3.4% for the furmonertinib arm.
In conclusion, furmonertinib demonstrated improved efficacy and a favorable safety profile when compared to gefitinib in patients with classical EGFR mutations in first-line NSCLC. The data especially highlighted the potential of furmonertinib to manage and prevent CNS metastases, suggesting an advantage over gefitinib in this patient subset. Importantly, the trial was designed for the first-line setting; therefore, extrapolating data to second-line or greater NSCLC treatment settings would require additional studies specifically in those populations.
FURVENT Phase 3 study
Study Summary:The FURVENT study is a global, Phase 3, randomized, multicenter, open-label trial that aims to evaluate the efficacy and safety of furmonertinib compared to platinum-based chemotherapy in patients with untreated, locally advanced or metastatic non-squamous NSCLC harboring EGFR exon 20 insertion mutations. Approximately 375 patients are expected to be enrolled and randomized in a 1:1:1 ratio to either furmonertinib 240 mg daily, furmonertinib 160 mg daily, or platinum-based chemotherapy (carboplatin or cisplatin, plus pemetrexed intravenously based on investigator's choice).
The study began in June 2023, with estimated primary completion by August 2025 and study completion by February 2028. The primary outcome measure is Progression-Free Survival (PFS) assessed by a blinded independent central review (BICR), with secondary measures including Overall Survival (OS), PFS by investigator assessment, Overall Response Rate (ORR), Duration of Response (DOR), Time to Second Progression-Free Survival (PFS2), CNS-specific outcomes, changes in quality-of-life questionnaires, and the safety and tolerability profile of furmonertinib, as well as plasma concentrations of furmonertinib and its major metabolite, AST5902.
The study design for the FURVENT clinical trial shows a rigorous approach by including two different dose levels of the investigational drug, allowing for comparison of dose-dependent effects and optimizing the therapeutic window. The use of platinum-based chemotherapy as a comparator is appropriate as it represents the current standard of care for such patients. The trial's measures include both efficacy and safety endpoints, which are important for a comprehensive assessment of furmonertinib’s potential as a first-line treatment.
However, being an open-label study, there could be potential biases as both the patients and the investigators are aware of the treatments being administered. This might influence patients' self-reporting of symptoms or side effects and could potentially affect the objectivity of the investigator assessments. The lack of blinding could also have implications for subjective endpoints, such as quality of life.
Operational and Technical Challenges
Conducting a global study introduces logistical complexities including variability in regulatory requirements, patient populations, standard of care across different countries, and the need for consistent training across multiple sites to ensure fidelity to the study protocol.
Further, as the study design includes brain metastases-specific outcomes, there might be challenges concerning the accurate and standardized assessment of these endpoints. CNS metastases are notoriously difficult to measure, requiring specialized imaging and expertise.
Also, there will be a need for robust mechanisms to manage and monitor the two oral doses of furmonertinib to avoid dosing errors.
The pharmacokinetic assessments will require careful timing and handling of blood samples to accurately measure plasma concentrations of furmonertinib and AST5902. These processes introduce an additional layer of complexity in terms of sample collection, storage, and transport to the analysis site.
Given the long duration of the study, maintaining patient engagement and minimizing loss to follow-up is also a potential operational challenge.
Primary and Secondary Endpoints
The primary endpoint, PFS determined by BICR, is an appropriate choice as it directly measures the effect of furmonertinib on halting disease progression, which is a critical consideration for patients and clinicians. BICR will add objectivity to assessments, which is especially important given the open-label design.
The secondary endpoints, including OS, ORR, DOR, PFS2, CNS-specific outcomes, changes in quality of life (via QLQ-C30 and QLQ-LC13), and safety and tolerability, are also well-suited. These endpoints will provide comprehensive data on furmonertinib's impact on various aspects of the disease, patient well-being, and potential side effects.
Inclusion / Exclusion Criteria
The inclusion criteria outline a population that will yield relevant data on the question at hand. Including patients who have a documented EGFR exon 20 insertion mutation ensures that the trial is assessing the drug in a population where it is expected to have a mechanism-based effect. Furthermore, the exclusion of patients who have had previous anticancer regimens for advanced or metastatic NSCLC reduces confounding variables that could affect the interpretation of the efficacy or safety of furmonertinib.
However, the requirement of a treatment-free interval of at least 12 months for those who have received neo-adjuvant or adjuvant therapy might limit the generalizability of the study findings. This criterion could exclude patients with more aggressive disease progression. Additionally, including patients with CNS metastases can increase the complexity of the study but is essential to understand the CNS activity of furmonertinib, which is a critical assessment area in NSCLC.
Potential risks
Reproducibility challenges might arise from variations in the documentation of EGFR exon 20 insertion mutations. If testing is not standardized across sites or if local testing allows for entry into the study, there could be variability in how patients are classified as mutation-positive. Furthermore, allowing both tissue and blood-based documentation of mutation status may introduce variability; tissue biopsies are often considered the gold standard, but blood-based tests (liquid biopsies) are less invasive and increasingly used in clinical practice.
Ensuring reproducibility would benefit from standardized testing protocols, preferably with central verification of mutation status to ensure uniformity in patient selection. However, central testing might not always be feasible in a global study and could reduce the speed of recruitment.
In addition, the study's global nature may introduce variability in standard-of-care practices across regions, which could potentially affect the reproducibility of the results. These potential challenges emphasize the necessity of stringent operational controls and protocol adherence to ensure data quality and consistency across study sites.
FAVOUR Phase 1b study
This clinical study is a Phase 1b, open-label, multi-center trial designed to investigate the initial efficacy and safety of Furmonertinib Mesilate in patients with locally advanced or metastatic Non-Small Cell Lung Cancer (NSCLC) who have EGFR exon 20 insertion mutations. The study aims to enroll 30 subjects, split into two groups: 20 who have been previously treated and 10 who are treatment-naïve. In terms of treatment, previously treated patients are randomly assigned to receive either 160 mg/day or 240 mg/day of Furmonertinib Mesilate, with each subgroup consisting of 10 patients. Meanwhile, all treatment-naïve patients will receive 240 mg/day. The primary measure of the study's success is the Objective Response Rate (ORR), while secondary outcomes include Disease Control Rate (DCR), Duration of Response (DOR), Depth of Response (DepOR), Progression-Free Survival (PFS), Overall Survival (OS), CNS ORR, the safety profile, and the pharmacokinetic (PK) profile of Furmonertinib Mesilate and its metabolites. The study is scheduled to run from August 2020 to December 2023, with a primary completion date estimated for March 2023.
However, there are several critiques of this study design. Being a Phase 1b study, its main objective is to assess safety and determine preliminary efficacy, but due to its small scale, it may not be sufficiently powered to confirm efficacy conclusively. The randomization process is limited to the previously treated cohort and involves two dosage levels, which, given the small sample size, may make it difficult to discern significant differences in efficacy and safety between the groups. With only 30 subjects, the study might not represent a diverse enough patient population to extend its findings to the broader NSCLC population with EGFR exon 20 insertion mutations. Moreover, the absence of a control group using standard treatment or a placebo limits the study's capacity to directly compare the effectiveness of Furmonertinib to current treatments. Finally, the open-label nature of the trial could introduce bias in the reporting of responses and the assessment of adverse events.
Operational and Technical Challenges
Recruitment: Identifying eligible NSCLC patients specifically with EGFR exon 20 insertion mutations may be challenging due to the rarity of this mutation.
Dosing Strategy: The use of two different doses in the treated and treatment-naïve populations might result in operational complexity and potential confusion during the implementation phase.
Assessment Accuracy: With an open-label design, the investigators' and participants' knowledge about the treatment could introduce bias in the assessment of subjective outcomes like side effects.
Standardization: Consistency in determining ORR, DCR, DOR, and other response measures across multiple centers is critical but may be challenging, requiring rigorous standardization and training.
Biomarker Analysis: Collection and analysis of peripheral blood ctDNA require careful handling, consistent protocols, and specialized equipment, which can be challenging on a multicenter scale.
Follow-Up: Keeping track of patients over time to gather data on long-term outcomes like OS and PFS can be difficult, especially with a small cohort where each participant's data carries significant weight.
Safety Monitoring: In early-phase trials, safety monitoring is of the utmost importance. The relatively high dosage levels of an experimental drug warrant close oversight for any adverse effects.
Data Interpretation: The small sample size demands cautious interpretation of the study results. With limited statistical power, the study is mainly explanatory and may not be definitive in establishing Furmonertinib's efficacy and safety profile.
Interim results
Summary of Clinical Data Supporting Furmonertinib in First-line NSCLC with EGFR Exon 20 Insertion Mutations:
The FAVOUR study is a Phase 1b clinical trial in China evaluating furmonertinib in patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) harboring EGFR exon 20 insertion mutations. The study consists of three cohorts with 30 patients each: treatment-naïve patients and two groups of previously treated patients, receiving either 160 mg or 240 mg of furmonertinib daily.
Clinical Results (as of June 15, 2023):
Patient Population: 86 enrolled, with 80 having measurable disease at baseline.
Efficacy:
- The treatment-naïve patient group showed a confirmed objective response rate (ORR) of 79%.
- Previously treated patients had an ORR of 46% for the 240 mg cohort and 39% for the 160 mg cohort.
- Median duration of response (DOR) was 15.2 months for treatment-naïve patients and 13.1 months and 9.7 months for the previously treated 240 mg and 160 mg cohorts, respectively.
- Median progression-free survival (PFS) was 10.7 months for treatment-naïve patients, and 7.0 and 5.7 months for the 240 mg and 160 mg previously treated cohorts, respectively.
- Disease control rate (DCR) was 100%, 92%, and 85% across the respective cohorts.
Interim median tumor size reductions were 51%, 54%, and 36% across treatment-naïve, 240 mg pretreated, and 160 mg pretreated cohorts respectively.
Safety:
- Generally well tolerated, with safety results similar to prior trials.
- Treatment-related serious adverse events (TRSAEs) included abnormal hepatic function, decreased platelet count, abnormal uterine bleeding, interstitial lung disease, diarrhea, and others.
- 21 treatment-emergent serious adverse events (TESAEs) were observed.
- Grade 3 or higher TRAEs occurred in 13% of treatment-naïve, and 18% and 29% of previously treated patients at the 160 mg and 240 mg doses, respectively.
- Dose interruptions occurred in 14%, 32%, and 23% of the respective groups, while dose reductions due to TRAEs were reported in 13% of naïve patients, and 11% and 18% of the previously treated 160 mg and 240 mg cohorts.
Only two patients discontinued the trial due to TRAEs.
Summary:
- Furmonertinib led to significant tumor size reductions across all patient cohorts.
- Rapid responses were observed, with most having a partial response by the first assessment at six weeks.
Final reporting, including the overall survival (OS) data, is expected to be released in 2024. The current clinical data demonstrates that furmonertinib is effective and relatively well-tolerated as a first-line treatment for NSCLC patients with EGFR exon 20 insertion mutations. However, interim data is subject to confirmation upon ongoing trial evaluation.
FURTHER Phase 1b study
Summary of Study Design:The study is a Phase 1b, open-label, multi-center, dose-escalation, and dose-expansion study of furmonertinib in advanced or metastatic NSCLC patients with activating EGFR or HER2 mutations, including less common ones. The purpose is to evaluate the safety, pharmacokinetics, and preliminary antitumor activity of the drug. About 170 participants are expected to be enrolled across two stages: Stage 1 involves dose escalation to determine the maximum tolerated dose (MTD) and any dose-limiting toxicities (DLTs), with additional patients included in backfill cohorts; Stage 2 is the dose expansion, where more patients are treated at the established dose for further assessment. This will include different cohorts based on the type of EGFR or HER2 mutation present.
- Critique of the Study Design:
- Phase 1b Nature: Being an early-phase trial, the primary focus on safety and pharmacokinetics is appropriate. However, the findings concerning antitumor activity should be considered preliminary.
- Dose Escalation: The dose-escalation approach is a standard and effective method for finding the MTD, but it requires careful monitoring and may involve a risk of under-treatment or over-treatment for some participants.
- Dose Expansion: Expanding patient cohorts at an identified dose level helps to further assess the safety profile and provides initial efficacy data, which can guide phase 2 dose selection.
- Non-randomized and Open-label: The study is non-randomized and open-label, which is typical for early-phase studies but may introduce bias due to the lack of a comparator group or blinding.
- Sequential Assignment: This approach is suitable for Phase 1 studies where a new treatment is being tested for the first time in humans, but it doesn't offer the comparative data one might receive from parallel-group designs.
- Mutation-specific Cohorts: The study's division into specific mutation cohorts will provide more precise information on the drug's activity across different genetic subtypes of NSCLC.
- Operational and Technical Challenges:
- Patient Recruitment: Identifying and recruiting a sufficient number of patients with specific activating mutations, including less common ones, may be challenging.
- Safety Monitoring: The study involves risk due to the untested nature of the drug and the need for close monitoring for adverse effects, particularly in the dose-escalation phase.
- Pharmacokinetic Studies: Detailed PK studies are technical and resource-intensive but are necessary for understanding the drug's metabolism and distribution.
- Efficacy Assessment: Preliminary efficacy findings in Phase 1b trials can vary significantly and may not be predictive of later-phase trial outcomes.
- Regulatory Compliance: Complying with regulatory requirements across multiple centers can be complex, notably in an open-label study where strict adherence to the protocol is essential for ensuring the quality and reliability of the data.
The design's focus on a specific patient population with activating mutations should generate valuable data about furmonertinib's activity in these subgroups. However, the results may not be generalizable to the broader NSCLC population, and larger, randomized, controlled trials will be needed to confirm efficacy and safety before furmonertinib can become a standard care option for these mutations.
Potential for Proof-of-Concept:The potential for this study to provide proof-of-concept for the use of furmonertinib in NSCLC with EGFR PACC mutations is high given the targeted approach and patient selection criteria. The proof-of-concept could be established through the demonstration of safety as well as preliminary efficacy including tumor response, stabilization of the disease, or other clinical benefits in patients carrying these specific mutations.
The endpoints chosen are appropriate for the study's phase and objectives. They are designed to confirm that furmonertinib can be safely administered to patients and provide the initial data on its potential efficacy against NSCLC with a particular focus on EGFR PACC mutations.
Inclusion / Exclusion Criteria:The inclusion criteria are designed to capture a specific patient population that could most benefit from furmonertinib:
- Patients with advanced or metastatic NSCLC, ensuring that the study focuses on a group for whom the condition is not amenable to curative surgery or radiotherapy.
- The need for disease progression after standard therapy or for standard therapy to be ineffective or intolerable selects patients who require new therapeutic options.
- The specific inclusion of patients with various forms of EGFR or HER2 mutations allows for the targeted assessment of furmonertinib's efficacy against these mutations.
However, these stringent inclusion criteria may lead to challenges in recruiting enough participants, given the rarity of some of the specified mutations.
Exclusion Criteria:The exclusion criteria are equally well-defined, aiming to minimize confounding factors that could affect the study's outcomes. The exclusion of patients who have recently received other cancer therapies ensures that the effects of furmonertinib can be attributed directly to the drug, rather than to lingering effects of previous treatments.
- Operational and Reproducibility Challenges:
- Enrollment Challenge: Recruiting a sufficient number of patients with the specified mutations, especially rarer ones, might be difficult and could potentially extend the recruitment period and geographic scope of the study.
- Mutation Testing: Validated results are required for confirming the mutations, which requires access to high-quality laboratory testing. Reproducibility may be affected by the variability in mutation testing accuracy across different labs or regions.
- Patient History: The requirement that patients have disease progression after standard therapy or that therapy was intolerable ensures the inclusion of a specific patient set but may exclude patients with different treatment histories, affecting the applicability of the study findings.
- Prior Treatments: Excluding patients with recent cancer therapy necessitates strict documentation and adherence to washout periods, which requires patient cooperation and accurate record-keeping.
Overall, the study design's inclusion and exclusion criteria align well with its aim to evaluate furmonertinib's utility for first-line or greater NSCLC treatment with EGFR PACC mutations. However, the stringent criteria likely reduce the potential study population size, potentially affecting the speed of enrollment and potentially the reproducibility and generalizability of the study findings.
SHP2 combination Phase 1b study
Summary of Study Design
The Phase 1b trial is focused on assessing the safety, pharmacokinetics, and preliminary efficacy of a combination therapy consisting of furmonertinib and a SHP2 inhibitor, ICP-189, in patients with EGFR-mutant (EGFRm) NSCLC that have classical mutations. The rationale for this combination stems from the role of SHP2 in EGFR signaling pathways and in the development of resistance to EGFR tyrosine kinase inhibitors (TKIs) via pathways such as MET signaling. Preclinical models have suggested that SHP2 inhibitors can potentiate the activity of EGFR TKIs and help overcome resistance mechanisms.
Enrollment for the study is anticipated to begin in 2024, with an expansion cohort planned for patients previously treated with a third-generation EGFR TKI once an appropriate dose and schedule have been determined. The study aims to provide potential proof of concept for second-line treatment of classical EGFRm NSCLC by 2026 and for first-line therapy in EGFR-TKI naïve patients by 2027.
Critiques of the Study Design
Study Timeline: The study may face challenges in keeping on the proposed timelines, especially given the complexity involved in combination therapies.
Pharmacokinetics (PK) and Pharmacodynamics (PD): While the study is designed to evaluate PK, it may also benefit from incorporating pharmacodynamics endpoints to understand how the drug affects the target pathway and whether inhibiting SHP2 produces the desired biological effect.
Efficacy Assessment: Preliminary efficacy in Phase 1b trials is challenging because they are typically designed for safety and dosing. Establishing efficacy usually requires a larger population and randomized trials.
Patient Population: The strategy to start with patients previously treated with a third-generation EGFR TKI and then proceed to a TKI-naïve cohort offers a staged approach to development. Still, it may face difficulties in recruiting the right patient populations, given that the trial is mutation-specific and involves previously treated patients.
Resistance Mechanisms: While the combination is aimed to overcome resistance, the specific mechanisms of resistance in the study population should be well-defined for the findings to be meaningful.
Operational or Technical Challenges
Recruitment: Identifying and recruiting patients with classical EGFR mutations who have been previously treated with third-generation EGFR TKIs and are suitable for the study can be challenging and time-consuming.
Combination Therapy: Managing the safety and efficacy profiles of two drugs combined can be complex, especially for dose-limiting toxicity assessments.
Biomarker Testing: Consistent and accurate biomarker testing is needed to identify appropriate patients and may require standardized testing protocols across different sites.
Clinical Timeline: Given the stated anticipation of proof-of-concept results in 2026 and 2027, the study may face pressures to complete the necessary steps quickly, which can be difficult with the careful dose evaluations and patient monitoring required in Phase 1 studies.
Drug Interaction and Safety Monitoring: As with any novel combination therapy, there is the potential for unexpected drug interactions, necessitating vigilance in monitoring and reporting of adverse events.
Data Management and Analysis: With the incorporation of both safety and preliminary efficacy data, as well as PK information, the data analysis plan must be robust to handle the intricacy of data and provide meaningful outcomes.
Overall, the study design is innovative in its aim to overcome resistance to EGFR TKIs in NSCLC patients with classical EGFR mutations. However, there are potential challenges in recruitment, study timelines, and the complexity of managing combination therapies that will have to be addressed to ensure the successful completion of the trial and meaningful results.Without the specific inclusion and exclusion criteria for the study, I can only provide a general commentary on the potential of the study to provide proof-of-concept for the use of furmonertinib in second-line or greater NSCLC with classical EGFR mutations, as well as discuss some general considerations regarding study design and possible reproducibility challenges.
Potential for Proof-of-Concept
The design of a Phase 1b clinical trial combining furmonertinib with a SHP2 inhibitor, such as ICP-189, for NSCLC with classical EGFR mutations holds significant promise for several reasons:
Targeted Therapy: By using a combination therapy that targets both the EGFR mutation and downstream signaling components such as SHP2, the treatment has the potential to address both primary tumor signaling and bypass pathways that can lead to TKI resistance.
Resistance Management: Second-line therapies in NSCLC often aim to tackle drug resistance that has developed following first-line treatments. Exploring SHP2 inhibition as a means to potentiate EGFR inhibition may offer a new avenue for patients who have developed resistance to initial therapies.
Synergistic Potential: Given the preclinical evidence that SHP2 inhibitors can enhance the efficacy of TKIs and overcome resistance, the study holds potential to demonstrate a proof-of-concept for the therapeutic synergy between furmonertinib and SHP2 inhibitors.
Appropriateness of Primary and Secondary Endpoints
For Phase 1b trials, the primary endpoints typically focus on safety and tolerability to determine the appropriate dose for subsequent studies. Pharmacokinetics will also be a key primary endpoint, assessing how the body processes the drugs when administered together. Preliminary efficacy, often a secondary endpoint at this stage, provides early evidence on whether the drug combination is having the desired effect on tumor growth.
For this proof-of-concept study:
Safety and Tolerability: This ensures that the drug combination does not result in prohibitive toxicity.
Pharmacokinetic Profile: Understanding the PK of the drugs helps in optimizing dosing schedules and potential interactions between the two drugs.
Preliminary Efficacy: Indications of efficacy, even in a Phase 1b trial, are crucial for determining whether the combination is worth pursuing in larger trials.
Inclusion / Exclusion Criteria
Though the specific criteria are not provided, here are typical considerations that would likely be part of the criteria:
Specific Mutations: Patients would need to have confirmed classical EGFR mutations, which the drugs are meant to target.
Previous Treatment: Since this would be for second-line therapy or greater, patients would likely have had to fail a first-line treatment or developed resistance to a prior EGFR TKI.
Performance Status: Generally, patients would need to have a certain level of overall health or performance status to tolerate the investigational therapy.
Reproducibility Challenges
Patient Selection: If inclusion criteria are too narrow, it may be difficult to recruit enough participants and later reproduce the study in a broader patient population.
Complex Regimens: Combination treatments with specific schedules might be challenging to reproduce exactly in real-world settings or in follow-up studies.
Mutation Diversity: Even among 'classical' EGFR mutations, there can be variability in the mutations present, potentially leading to variable responses to treatment.
In summary, the study holds significant potential to establish a new treatment paradigm for NSCLC patients with classical EGFR mutations, especially in the context of second-line therapy. The main goals will be determining the safety and pharmacokinetic profiles of the drug combination and gaining preliminary data on efficacy. The reproducibility challenges will largely depend on the specifics of the inclusion and exclusion criteria, which would need to balance the desire for a homogeneous study population with the need for results that are applicable to the broader population of NSCLC patients.
Clinical Data Results:
- Pharmacokinetic data as of June 15, 2023, is consistent with previous data from the FAVOUR trial, showing similar steady-state levels of furmonertinib and its major metabolite, AST5902.
- Adverse events (AEs) are consistent with those typically associated with EGFR-targeted TKIs, with the most common being diarrhea, stomatitis, and rash. Frequency of dose reduction and discontinuation due to AEs remains low, suggesting the drug is tolerable.
- Preliminary efficacy data shows a reduction in tumor size, with confirmed and unconfirmed partial responses in NSCLC patients with EGFR exon 20 insertions.
Significance:
- The preclinical data suggested that furmonertinib could be effective against a broader number of PACC mutations compared to the currently available treatment option, afatinib, which has data supporting its use in only two of the PACC mutations.
- The FURTHER trial is still enrolling, and robust engagement with medical oncology centers around the globe is ongoing to identify and enroll the right patient population.
Weekly analyses of biotech startups, generated by AI
Receive high quality, AI-generated analyses of biotech startups, public companies and scientific papers each week.
Market overview
First-line or greater NSCLC with EGFR PACC mutations
The market opportunity for Furmonertinib in first-line NSCLC with EGFR Exon 20 insertion mutations could be significant, as it targets a subset of NSCLC patients with a specific genetic mutation that is associated with a poorer prognosis and fewer effective treatment options. Here's an assessment based on the current landscape of NSCLC treatment:
-
Prevalence and Need: NSCLC constitutes the majority of lung cancer cases, with a significant portion of these cases presenting with EGFR mutations. While the incidence of EGFR Exon 20 insertion mutations is less common compared to classical EGFR mutations, they still represent a meaningful patient population given the high incidence of lung cancer. Since patients with these mutations tend to have a worse prognosis, there is a high unmet medical need for effective treatments.
-
Current Standard of Care: For advanced NSCLC patients with classical EGFR mutations, the standard of care includes EGFR tyrosine kinase inhibitors (TKIs) such as gefitinib, erlotinib, afatinib, and osimertinib. Osimertinib in particular has shown effectiveness in the first-line treatment of NSCLC with classical EGFR mutations. However, these therapies often have limited efficacy in patients with EGFR Exon 20 insertion mutations, leaving a gap in the treatment options for these patients.
-
Market Opportunities and Challenges: The opportunity for Furmonertinib lies in its potential to serve as an effective treatment for the subset of NSCLC patients harboring the Exon 20 insertion mutations. If clinically successful, it could address the unmet need and gain a substantial market share among this patient population. However, the efficacy and safety profile compared to other treatments will be crucial, as well as pricing and market access strategies.
-
Competing Treatments: The market opportunity should also be assessed in the context of competitive products. Amivantamab (Rybrevant) by Janssen and mobocertinib (Exkivity) by Takeda are therapies approved by the FDA for patients with NSCLC harboring EGFR Exon 20 insertion mutations who have received prior platinum-based chemotherapy. However, Takeda in October 2023 voluntarily withdrew mobocertinib from the market for treating patients with Exon 20 mutations due to failure to meet the primary endpoint of its confirmatory Phase 3 trial. Any new drug like Furmonertinib would need to demonstrate improved outcomes or other advantages over these existing therapies.
-
Diagnostic Testing: The availability and adoption of FDA-approved DNA testing for EGFR mutations, including exon 20 insertions, will influence the market opportunity. Widespread use of such tests could facilitate early and precise identification of patients who may benefit from Furmonertinib, thereby optimizing its market reach.
In summary, while there is a significant opportunity for Furmonertinib in treating NSCLC patients with EGFR Exon 20 insertion mutations, the product’s success will depend on its ability to demonstrate safety and efficacy, regulatory approvals, competitive differentiation, favorable pricing and reimbursement, and breakthroughs in patient identification through genetic testing. Successful penetration of this market will require strategic positioning alongside or against established treatments in what is increasingly becoming a personalized treatment landscape for NSCLC.
Several promising treatments for first-line non-small cell lung cancer (NSCLC) with EGFR Exon 20 insertion mutations could potentially compete with Furmonertinib:
-
Amivantamab (Rybrevant): Developed by Johnson & Johnson, amivantamab is an anti-EGFR and anti-MET bispecific antibody that has shown promise in the treatment of NSCLC with EGFR exon 20 insertion mutations. Its performance in the Phase 3 PAPILLON study indicates a potential competitive edge, especially since it met its primary endpoint as mentioned.
Sunvozertinib (DZD9008): Developed by Dizal Pharmaceutical, sunvozertinib is an oral EGFR inhibitor currently being evaluated in a Phase 1 clinical trial for first-line NSCLC patients with EGFR exon 20 insertion mutations. As a targeted therapy, sunvozertinib could be a direct competitor to Furmonertinib if it demonstrates favorable safety and efficacy profiles.
ORIC-114: This is another oral EGFR inhibitor developed by Oric Pharmaceuticals. Like Sunvozertinib, ORIC-114 is undergoing a Phase 1 trial in first-line NSCLC patients with EGFR exon 20 insertion mutations. Its effectiveness in this trial could make it a viable alternative to Furmonertinib.
Drugs from Major Pharma: Companies such as AstraZeneca, Blueprint Medicines Corp, Taiho Pharmaceutical, Boehringer Ingelheim, and Bayer AG have substantial resources and a history of successful drug development. Any new molecules or combination therapies emerging from these companies could change the competitive landscape.
Partnerships and Collaborations: Smaller biotech firms are collaborating with larger pharmaceutical companies to pool resources, knowledge, and technology. If successful, these partnerships could lead to the rapid development of new drugs that might outperform or undercut Furmonertinib.
Generics and Biosimilars: As patents expire on existing EGFR-targeted therapies, generics and biosimilars may become available at a reduced cost, providing economic options for treatment and affecting the market share of newer, branded drugs.
Key competitive factors for Furmonertinib will likely be its clinical efficacy, safety profile, ease of administration, and overall cost compared to these potential competitors. Successful marketing and securing reimbursement from healthcare providers will also be critical, as will navigating regulatory pathways to ensure timely market entry. Keeping abreast of the latest clinical trials and the progress of these competing drugs will be essential for gauging the future market outlook for Furmonertinib.
Ultimately, the exact role that Furmonertinib might play in treating NSCLC with EGFR Exon 20 insertion mutations will largely depend on data from ongoing or future clinical trials that prove its efficacy and safety, cost-effectiveness, as well as its place in relation to existing and other emerging therapies.
First-line or greater NSCLC with EGFR PACC mutations
Furmonertinib could represent a significant market opportunity in the treatment of non-small cell lung cancer (NSCLC) with EGFR PACC mutations, for the following reasons:
-
Unmet Medical Need: EGFR PACC mutations are categorized among the less common EGFR mutations in NSCLC, which have historically been associated with poorer prognosis and limited treatment options compared to classical EGFR mutations. There is an unmet medical need for treatments that are more effective for these subtypes.
-
Targeted Patient Population: PACC mutations, much like exon 20 insertions, represent a specific genetically-defined subgroup of NSCLC. The ability to precisely target such mutations with a therapy like Furmonertinib could lead to better clinical outcomes for these patients.
-
Standard of Care: Currently, the standard of care for first-line treatment of NSCLC with EGFR mutations largely includes EGFR tyrosine kinase inhibitors (TKIs) such as erlotinib, gefitinib, afatinib, and osimertinib tailored towards classical EGFR mutations. However, patients with PACC mutations may not respond as well to these existing therapies, highlighting a gap that Furmonertinib might be able to fill.
-
Market Dynamics: With the increasing use of genetic profiling in cancer treatment and the development of more advanced diagnostics like FDA-approved next-generation sequencing (NGS) tests, more patients with rare mutations could be identified, increasing the potential market for targeted therapies like Furmonertinib.
-
Competition: Although there is a subset of drugs approved for NSCLC with classical EGFR mutations, and more recently for EGFR exon 20 insertion mutations, such as Rybrevant (amivantamab) and Exkivity (mobocertinib), there are fewer options for patients with PACC mutations. Therefore, a product showing efficacy in this area could quickly become a preferred option if supported by substantial clinical data.
-
Clinical Development: The successful clinical development and subsequent approval of Furmonertinib—demonstrating safety and efficacy in treating PACC mutation-positive NSCLC—would be a key factor in determining its market potential.
-
Regulatory Pathway: There may also be expedited regulatory pathways (such as breakthrough therapy designation, accelerated approval, or orphan drug designation) available for therapies that address rare and serious conditions with few or no therapeutic options. Such pathways could facilitate a faster route to market for Furmonertinib.
In conclusion, the potential market opportunity for Furmonertinib in treating NSCLC with EGFR PACC mutations is underscored by the lack of specifically-targeted treatments for this subgroup, the overall high morbidity and mortality of lung cancer, and the rising precision oncology approach that favors targeted therapies. The drug's actual market impact will depend on the results of clinical trials, the competitive landscape, and how emerging therapies are integrated into oncology treatment protocols.
There are several in-development therapies for first-line or greater NSCLC with EGFR mutations, including PACC mutations, that could present competition for Furmonertinib:
-
Amivantamab (Rybrevant): Johnson & Johnson's bispecific antibody targets EGFR and MET, and, although initially approved for EGFR exon 20 insertion mutations post-platinum therapy, its exploration in combination with chemotherapy in the Phase 3 PAPILLON study suggests potential broadening of its use. Depending on the outcomes of ongoing and future studies, amivantamab could be an option for patients with various EGFR mutations, potentially including PACC mutations.
Sunvozertinib: Dizal Pharmaceutical's oral EGFR inhibitor, which has been updated in clinical trials for EGFR exon 20 insertion mutations, could also be studied in PACC mutations if its mechanism of action is applicable. Its development status in first-line NSCLC could place it in direct competition with Furmonertinib.
ORIC-114: Oric Pharmaceuticals' EGFR inhibitor is currently in Phase 1 studies for patients with EGFR exon 20 insertion mutations, and could similarly be examined in PACC mutation-positive patients. Its progress may predict future competition with Furmonertinib directly for the same subset of patients.
Drugs from Established Pharma Companies: Many large pharmaceutical companies such as AstraZeneca, Blueprint Medicines, Taiho Pharmaceutical, Boehringer Ingelheim, and Bayer have the resources to fast-track development, expand indications of existing EGFR inhibitors, or innovate new therapies for EGFR mutations. Their extensive capabilities in research, development, and marketing could present significant competition.
Partnerships: Collaborations between small biotechs and larger pharma may result in the discovery and development of novel therapies or combination treatments that could compete with Furmonertinib, especially if they yield more efficacious or durable responses.
Innovative Approaches: Companies are continually researching new methodologies, such as combination therapies, next-generation sequencing to identify patients, or targeted delivery systems, which could lead to new competitors in NSCLC treatment regimens.
For Furmonertinib to carve out a space for itself as a treatment for NSCLC with EGFR PACC mutations, it will need a differentiated clinical profile—a balance of safety, efficacy, and cost-effectiveness. How well it performs in clinical trials compared to these emerging competitors, its ease of use for patients (e.g., oral bioavailability vs. intravenous administration), and the ability of the developing company to secure regulatory approvals and favorable reimbursement will be critical. Additionally, the rapid pace of biopharmaceutical R&D and potential future mergers or acquisitions could further shape the competitive landscape.
Treating first-line non-small cell lung cancer (NSCLC) with EGFR PACC (posterior auricular canal closure) mutations with targeted therapies can be challenging, as these specific mutations are considered uncommon and research in this area is relatively less advanced as compared to treatments targeting classical EGFR mutations.
However, the following are notable treatments for NSCLC that may have relevance to patients with various EGFR mutations, including potentially those with PACC mutations:
Osimertinib (Tagrisso): A third-generation EGFR tyrosine kinase inhibitor (TKI) developed by AstraZeneca. It is designed to target T790M resistance mutation in EGFR-positive NSCLC but is also effective against classical EGFR mutations. Its efficacy for PACC mutations specifically may not be established, but its broad activity against various EGFR mutations makes it a significant drug in the NSCLC treatment landscape.
Erlotinib (Tarceva) and Gefitinib (Iressa): These are first-generation EGFR TKIs used to target classical EGFR mutations in NSCLC. While they are not specifically indicated for PACC mutations, they highlight the evolution of EGFR-targeted therapy in NSCLC.
Afatinib (Gilotrif): Another EGFR TKI, from Boehringer Ingelheim, is a second-generation drug that has broader activity against various EGFR mutations in NSCLC. Again, while not specifically for PACC mutations, it is part of the growing class of TKIs used in NSCLC treatment.
Amivantamab (Rybrevant): This is an EGFR/MET bispecific antibody by Johnson & Johnson that received FDA approval for patients with NSCLC harboring the EGFR exon 20 insertion mutations. It represents a novel approach beyond traditional TKIs.
Mobocertinib (Exkivity): Developed by Takeda, this oral TKI specifically designed for patients with EGFR exon 20 insertion mutations has been granted accelerated FDA approval for use after platinum-based chemotherapy fails. It exemplifies focused treatment advancements for specific EGFR mutation subtypes.
As of early 2023, there may not be any drugs specifically approved for EGFR PACC mutations, stressing the importance of continued research and development in this area. A drug like Furmonertinib could potentially fill this gap if clinical trials demonstrate efficacy and safety for patients with these specific mutations.
Here's how Furmonertinib might fit into the current treatment landscape for NSCLC with EGFR PACC mutations:
Targeted Therapy Niche: If Furmonertinib shows significant efficacy against PACC mutations, it could fulfill a currently unmet need in the NSCLC therapeutic arena. Since these mutations are less common and current EGFR TKIs are primarily focused on the more prevalent EGFR mutations, Furmonertinib could be positioned as a specialized treatment for this subgroup.
Potential First-line Treatment: Depending on clinical trial results, Furmonertinib could either become a first-line treatment or a second-line therapy post progression on standard chemotherapy or other EGFR TKIs, contingent upon its effectiveness and tolerability profile in comparison to standard care.
Clinical Trial Data: The extent to which Furmonertinib can penetrate the standard of care will largely be driven by clinical trial data. This includes data not only on the effectiveness of the drug but also on its safety profile, dosage optimization, and any potential side effects or resistance patterns.
Combinatorial Approaches: The current trend in oncology is moving toward combination therapies to prevent resistance and improve patient outcomes. If Furmonertinib has a mechanism that synergizes with other existing treatments, such as chemotherapy, immunotherapy, or other targeted therapies, it might be used within a combination regimen.
Competition and Differentiation: Furmonertinib's potential role in the standard of care will also be determined by how it compares to other treatments targeting PACC mutations, whether those therapies are already approved, are contemporaneous, or come to market later.
Market Access and Acceptance: Finally, physician acceptance, market access, insurance coverage, and cost will influence Furmonertinib's place in the treatment of NSCLC with PACC mutations. To be adopted, the drug will need to demonstrate not just clinical benefit but also cost-effectiveness.
Therefore, while Furmonertinib could potentially be a valuable addition to the NSCLC treatment paradigm, especially for patients with EGFR PACC mutations, many factors including robust clinical trial data, regulatory approval, cost vs. benefit analysis, and strategic positioning against competitors will all influence its adoption and use as part of the standard of care.
Second-line or greater NSCLC with classical EGFR mutations
The market opportunity for Furmonertinib in second-line or greater NSCLC with classical EGFR mutations can be summarized and explored in the following aspects:
High Incidence of NSCLC and EGFR Mutations: Lung cancer remains a leading cause of cancer deaths worldwide, with NSCLC making up the largest proportion of cases. Among NSCLC patients, a significant portion, particularly those from Asian countries, non-smokers, women, and younger adults, harbors classical EGFR mutations, which typically include exon 19 deletions and L858R mutations in exon 21. This high incidence presents a substantial market for EGFR-targeted therapies.
Existing Therapies and Resistance: The current standard of care for first-line treatment in NSCLC patients with classical EGFR mutations includes EGFR tyrosine kinase inhibitors (TKIs) like erlotinib, gefitinib, afatinib, and the more recent osimertinib, which is effective against T790M resistance mutations as well. However, most patients eventually develop resistance to these therapies, leading to disease progression and a need for second-line therapeutic options.
Unmet Medical Need in Second-Line Treatment: Once resistance to first-line EGFR TKIs occurs, treatment options become more limited, indicating an unmet medical need for effective second-line treatments. While chemotherapy and immunotherapy are options, targeted therapies that overcome resistance could present improved outcomes for patients.
Market Opportunity for Furmonertinib: If Furmonertinib proves to be efficacious and safe in overcoming resistance to first-line EGFR TKIs or offers benefits such as fewer side effects or improved quality of life, it could capture a significant market share as a second-line treatment. Its success will also depend on how it compares to other second-line treatments, including chemotherapy, immunotherapy options (e.g., PD-1/PD-L1 inhibitors), and other targeted therapies under development.
Competition: To fully understand the market potential for Furmonertinib, one must consider the competitiveness of the space. It would face direct competition from other drugs that offer treatment post-TKI resistance, such as the third-generation TKI osimertinib, which is already approved for patients with T790M mutations. Further, other drugs in development that target EGFR mutations after progression on the initial EGFR TKI, such as newer generation EGFR TKIs and combination therapies, also constitute competition.
Accessibility and Approval: For Furmonertinib to be integrated into the market successfully, it would need to secure regulatory approval based on data from rigorous clinical trials demonstrating its efficacy and safety as a second-line treatment. Additional factors including drug pricing, reimbursement policies, and global marketing strategies would also play roles in its commercial success.
In summary, Furmonertinib might have a notable market opportunity in the second-line or greater treatment of NSCLC with classical EGFR mutations if it demonstrates clinical benefits over existing therapies, specifically addressing the issue of resistance that arises with first-line EGFR TKI treatments. The extent of this opportunity would also depend on the rapid identification and confirmation of appropriate patients through FDA-approved diagnostic tests as treatment with targeted therapies becomes increasingly personalized based on the molecular profile of tumors.
In the competitive arena of second-line or greater treatments for NSCLC with classical EGFR mutations, several promising therapies are in development that may compete with Furmonertinib:
Osimertinib (Tagrisso) by AstraZeneca: Originally approved as a second-line treatment for patients with T790M mutation after progression on EGFR TKIs, osimertinib has now been moved to the first-line setting for patients with classical EGFR mutations due to its efficacy. Its role in second-line treatment remains robust, and continuous development could lead to newer generations or formulations.
Amivantamab (Rybrevant) by Johnson & Johnson: While currently approved for NSCLC patients with EGFR exon 20 insertion mutations, its successful Phase 3 PAPILLON study in first-line treatment suggests potential future applications or combination strategies that might also prove effective in second-line settings for classical EGFR mutations post-TKI therapy.
Dizal Pharmaceutical and Oric Pharmaceuticals: Both companies are working on oral EGFR inhibitors, sunvozertinib and ORIC-114 respectively, which are in clinical trials for NSCLC patients with EGFR exon 20 insertion mutations. Depending on their spectrum of activity, these drugs could potentially be studied and developed for use in classical EGFR mutation-positive NSCLC in the second-line setting.
Blueprint Medicines Corp, Black Diamond Therapeutics, Inc., Taiho Pharmaceutical Co., Ltd., Boehringer Ingelheim, and Bayer AG: These companies are also working on novel oncology therapeutics and could have undisclosed projects or early-phase research targeting EGFR mutations. Their financial resources and experiences in drug development position them as strong competitors.
Partnerships and Collaborations: Small biotech firms entering into partnerships with large pharmaceutical companies can result in acceleration of drug development and novel combination therapies which may address resistance mechanisms post-first-line therapy, presenting potential competition to Furmonertinib.
Additionally, with the advancements in biotechnology, it is likely that there will be:
New Targets and Combination Therapies: As scientific understanding of the mechanisms of resistance to current EGFR TKIs advances, new targets are being identified and targeted by novel agents. Combination therapies, including those that may integrate TKIs with immunotherapeutics or other targeted agents, are being explored to overcome resistance and improve outcomes.
Next-Generation TKIs: Further next-generation EGFR TKIs that can overcome a broader range of resistance mechanisms are under development and may offer improved specificity and tolerability compared to current therapies.
The market opportunity for Furmonertinib will be influenced by the ongoing evolution of these competitive products in terms of their efficacy, safety profile, convenience, and cost. To successfully establish itself, Furmonertinib will need to differentiate itself with a favorable balance of these factors and demonstrate clear clinical benefits in the second-line or greater treatment settings for patients with classical EGFR mutations. As with all experimental drugs, the actual competitive landscape will materialize following clinical trial results, regulatory approval processes, and market entry strategies.
Notable drugs used to treat second-line or greater non-small cell lung cancer (NSCLC) with classical EGFR mutations include targeted therapies that have been approved based on their ability to address resistance mechanisms that often develop after first-line treatment. Some of these therapies have also been recently approved and integrated into the range of treatment options for these patients.
Osimertinib (Tagrisso) – A third-generation oral EGFR tyrosine kinase inhibitor (TKI) developed by AstraZeneca specifically for patients with NSCLC who have developed the T790M mutation as a mechanism of resistance to first-generation EGFR TKIs. Osimertinib has demonstrated survival benefits and is now often used as a first-line treatment for EGFR-mutated NSCLC due to its efficacy.
Afatinib (Gilotrif) – A second-generation TKI that irreversibly inhibits EGFR (including common mutations like Exon 19 deletions and L858R mutations), developed by Boehringer Ingelheim. While initially used as a first-line treatment, it can also be considered for later lines of therapy, especially in different combinations or for patients who did not receive it initially.
Dacomitinib (Vizimpro) – This second-generation EGFR inhibitor from Pfizer is also an irreversible inhibitor of the EGFR tyrosine kinase. It is active against common EGFR mutations and has been used in the first-line setting, but remains a potential option for subsequent lines of therapy depending on individual patient treatment histories and the evolution of resistance.
Ramucirumab (Cyramza) – In combination with erlotinib, this angiogenesis inhibitor from Eli Lilly has been used for the first-line treatment of metastatic NSCLC with EGFR mutations based on the RELAY trial. While not directly targeting the EGFR TKI resistance, the combination strategy represents a newer therapeutic approach and may also have implications for later-line therapy settings.
Erlotinib (Tarceva) and Gefitinib (Iressa) – First-generation EGFR TKIs which have historically been utilized for first-line therapy but may still be used in subsequent lines of treatment, particularly when patients have not received them earlier or in certain clinical situations.
Recently approved therapies or those in late-stage development could also become noteworthy additions to the treatment options for second-line or greater NSCLC with classical EGFR mutations:
- Novel EGFR inhibitors that are being developed to overcome different resistance mechanisms.
- Combination therapies that include EGFR TKIs paired with agents targeting different pathways involved in tumor growth and survival, potentially circumventing or delaying resistance.
- Agents that target mechanisms of resistance beyond the T790M mutation are still needed, as are therapies that can address C797S or other complex mutation patterns that can arise after treatment with drugs like osimertinib.
It is essential to note that the treatment landscape for NSCLC is dynamic, with ongoing trials likely to introduce new therapeutic options, indications, and combination therapies. Due to the nature of cancer resistance mechanisms, alternative approaches that are less drug-specific but target the tumor's environment or leverage the patient's immune system are also becoming increasingly important to the treatment paradigm. These include, but are not limited to, checkpoint inhibitors or therapies directed at other molecular abnormalities. Always reference the most current clinical guidelines and FDA approvals for updated treatment options for NSCLC.
Furmonertinib's fit into the standard of care for second-line or greater treatment of NSCLC with classical EGFR mutations will largely depend on several key factors:
Treatment Efficacy: If Furmonertinib demonstrates superior or comparable efficacy to existing treatment options while offering benefits such as a better safety profile or longer duration of response, it can establish itself as a viable second-line or subsequent therapy for NSCLC patients with classical EGFR mutations.
Resistance Management: Second-line therapies for NSCLC are often focused on overcoming resistance to first-line EGFR TKIs. Furmonertinib could fit into the standard of care if it shows effectiveness against known resistance mutations such as T790M or provides a new mechanism to overcome or circumvent resistance.
Safety and Tolerability: The safety and side effect profile of Furmonertinib will also play a vital role. The drug will need to have tolerability that is at least comparable to current second-line therapies to be considered a new standard of care.
Patient Quality of Life: Improvements in the quality of life, such as fewer side effects, easier administration, or reduced treatment burden, could make Furmonertinib a preferred option for patients and clinicians.
Combinatorial Potential: The ability to combine Furmonertinib with other effective therapies, such as angiogenesis inhibitors or checkpoint inhibitors, could provide a more comprehensive approach to treatment, which could help it fit into a combination standard of care.
Cost-Effectiveness: Affordability and insurance coverage will impact its integration into the standard of care. If the drug can offer cost savings over other treatments either through lower pricing or by reducing the need for subsequent therapies, it could be more readily accepted by healthcare systems.
Regulatory Approval: Successful clinical trials resulting in regulatory approval for second-line or later use in classical EGFR mutation-positive NSCLC will be critical for Furmonertinib's adoption into standard of care. Positive phase III trial data and FDA approval would be necessary steps.
Should Furmonertinib meet these conditions and demonstrate clear benefits, it could fit into the current treatment algorithm either as a direct second-line treatment post-first-line EGFR TKI failure or as a subsequent option if resistance to second-line therapies develops.
The role of this drug will unfold as the results of clinical trials become public and as the drug goes through the regulatory approval process. Keeping abreast of emerging treatments, evolving patient populations, and real-world effectiveness data will further determine Furmonertinib's place in the treatment paradigm for NSCLC with classical EGFR mutations.
Revenue build and model
First-line or greater NSCLC with EGFR Exon 20 insertion mutations
Let's create a revenue build for Furmonertinib in the first-line treatment of NSCLC with EGFR Exon 20 insertion mutations. Please note these numbers are placeholders and will need to be adjusted with real-world data and market research:Prevalence of NSCLC with EGFR Exon 20 Insertion: For this, epidemiological studies would need to provide the incidence and prevalence rates. For hypothesis purposes, let's say 85% of the 228,820 new lung cancer cases are NSCLC, which gives us roughly 194,497 cases. If 5% of these harbor the EGFR Exon 20 insertions, we have approximately 9,724 patients eligible for treatment annually in the U.S.
Market Penetration: Considering all eligible patients may not be reached or diagnosed correctly, let's assume a peak market penetration of 45%.
Gross-to-Net Adjustments: Factoring in discounts, rebates, and other adjustments, let's assume a 40% reduction from the list price.
Insurance Coverage: Assuming 85% of treatments are covered by insurance at varying levels of copay and co-insurance.
Duration of Therapy: Based on clinical trial data showing the median duration of response and with adjustments for real-world use, let's assume an average of 9 months of continuous treatment per patient.
Price per Month of Therapy: A standard monthly cost for EGFR TKIs might range from $10,000 to $15,000, though pricing strategies would need to consider competitor pricing, healthcare system impact, and value-based pricing models.
Additional Considerations for Revenue Build:
- Annual Price Increases: Depending on the healthcare inflation rates and industry standards, a 2-4% price increase might be factored in.
- International Markets: Expansion into global markets will consider prevalence rates, market access, pricing, and insurance coverage in those areas.
- Competition Impact: Revenue may be affected by the entry of competing drugs or new clinical data impacting the use of Furmonertinib.
- Real-World Efficacy and Safety Data: Post-market surveillance could either positively or negatively impact the drug's use.
This revenue build is highly simplified and does not include specific market analyses, segmentation, or detailed financial modeling. Actual revenue projections would require a multidisciplinary approach involving market research, financial analysis, health economics, and in-depth knowledge of the competitive landscape and payer systems.
Probability of clinical success
To estimate probability of success, we can start with industry averages. Then we can adjust the estimates based on the specific qualities of the drug in question.
For Furmonertinib in particular, the Phase 2 data appears very promising. The high objective response rates (ORR), the duration of response (DOR), disease control rates (DCR), and interim median tumor size reductions indicate strong efficacy, especially in the treatment-naïve patient cohort. Additionally, the drug’s safety profile seems manageable, with a relatively low discontinuation rate due to treatment-related adverse events (TRAEs).
These positive results could suggest that Furmonertinib has a higher than average chance of progressing successfully through Phase 3 and onto FDA submission, assuming the Phase 3 trial design appropriately addresses the FDA’s requirements and continues to produce favorable efficacy and safety data.
To make a hypothetical estimate for the success of Furmonertinib, one might weigh the industry standard probabilities taking into account the relatively strong Phase 2 data. Given the strong Phase 2 data, we could hypothesize that the odds of Phase 3 success might be higher than the industry average. If we cautiously increase the probability by a factor reflective of the data, say by 10%, we might estimate Furmonertinib’s Phase 3 success as = 52.5%.
It is important to note these adjusted probabilities are still hypothetical; the actual chances of Furmonertinib’s success could differ significantly based on a multitude of factors that would be identified during a thorough risk assessment by the developing pharmaceutical company.
In practice, drug developers, investors, and other stakeholders often use more dynamic models and consider specific drug attributes, trial designs, current evidence, regulatory feedback, historical performance of similar drugs, and real-time updates from ongoing trials to refine such probability estimates.
First-line or greater NSCLC with EGFR PACC mutations
To calculate the hypothetical revenue build for Furmonertinib in the first-line or greater NSCLC with EGFR PACC mutations, we need to make several assumptions. Here are the steps for the revenue build:
- Incidence and Prevalence of NSCLC: 200,000 cases/year
Rate of EGFR PACC Mutations among NSCLC: 10%
Treatment Penetration and Market Share: Assume peak market penetration of 45%
Pricing: Assume the average treatment cost per patient per year for Furmonertinib: $180,000
Gross-to-Net Adjustments: 40%
Insurance Coverage: 80%
Duration of Therapy: 9 months
Please note, these are hypothetical placeholder estimates designed for the purpose of illustrating the revenue calculation process. Real numbers should be used based on actual market data, treatment regimens, pricing strategies, insurance coverage, and other relevant factors to obtain an accurate revenue forecast. Additionally, market uptake curves, competitive landscape shifts, and health economics should be analyzed to refine these projections over time.
To estimate the likelihood of Furmonertinib advancing through clinical trials, we can use the provided industry-standard success rates. However, it's important to note that the actual probabilities could differ based on a variety of factors including the strength of the preclinical data, the drug's mechanism of action, the target population, and the unmet medical need. Positive preclinical data may suggest a higher than average chance of success, but for the purpose of this response, we will use the standard rates provided.
Second-line or greater NSCLC classical EGFR mutations
To create a hypothetical revenue build for Furmonertinib in second-line or greater NSCLC with classical EGFR mutations, we would follow a multi-step process utilizing placeholder estimates. The following components would need considerations:
Annual NSCLC Incidence (ANI): The number of new NSCLC cases annually in the target market.
EGFR Mutation Prevalence (EMP): Percentage of NSCLC patients with classical EGFR mutations.
Second-Line Treatment Eligibility (STE): Proportion of patients with EGFR mutations eligible for second-line treatment.
Market Share of Furmonertinib (MSF): Projected market share captured by Furmonertinib for the relevant indication.
Average Treatment Cost (ATC): Annual cost of Furmonertinib treatment per patient.
Gross-to-Net Deduction (GND): Discount and rebate deductions from the list price.
Insurance Coverage Percentage (ICP): Estimated proportion of patients whose insurance covers Furmonertinib.
Average Duration of Treatment (ADT): The average length of time a patient remains on Furmonertinib.
Now, let's identify placeholder estimates for each item:
ANI: 235,000 (based on the estimated number of new lung cancer cases annually in the target market).
EMP: 30% (estimated percentage of NSCLC that would have classical EGFR mutations).
STE: 60% (proportion of patients failing first-line therapy and moving to second-line therapy).
MSF: 25% (market share captured by Furmonertinib, considering competition and drug efficacy).
ATC: $180,000 (annual cost of Furmonertinib per patient, which is a placeholder).
GND: 40% (assuming discounts, rebates, and other deductions).
ICP: 85% (assuming most patients will have insurance coverage for second-line therapy).
ADT: 8 months (considering the course of treatment for progression)
Please note that this is a highly simplified revenue model and does not account for various factors that could significantly affect actual revenues, including price fluctuations, market dynamics, variations in insurance coverage, patient dropout rates, patient adherence to therapy, competitive drug launches, and expanding indications. Additionally, economic and regulatory factors can impact market uptake and access to treatment. It also assumes that the duration of therapy and adherence rates do not change over time, which in reality may vary. Actual revenue projections require a detailed market analysis and dynamic forecasting models.
Platform overview
ArriVent BioPharma's scientific strategy appears to be centered around in-licensing and developing oncology drugs, with a particular emphasis on therapies that originated from China. They are looking to leverage the second-largest pharmaceutical market's growing research and development capabilities to introduce novel therapeutic programs to a global market. ArriVent aims to address the unmet medical needs of cancer patients through targeted approaches, and their initial lead asset is furmonertinib, a potential treatment for certain types of non-small cell lung cancer (NSCLC).
Scientific Strategy Overview:
Maximize the Potential of Furmonertinib: ArriVent is focusing on the development and commercialization of furmonertinib for treating EGFR mutation-positive NSCLC. Their goal is to use this asset to target both common and less common mutations, including exon 20 insertion mutations, for which there is a significant gap in effective first-line therapies.
Pursue Assets with a Clear Regulatory Path: They intend to leverage their extensive experience with regulatory agencies to streamline the path to approval for assets in development, thus ensuring a faster transition from clinical trials to market availability.
In-Licensing Strategy: The company aims to identify high-quality development candidates that have promising clinical data and then acquire the rights to these candidates outside of China. This means they could bring potentially efficacious treatments to other parts of the world where the therapies might not otherwise be available.
Focus on China as a Source for Novel Therapies: ArriVent believes that the wealth of biopharmaceutical R&D in China provides a ripe field for innovative therapies that could be brought to global populations. They plan to in-license and then develop these candidates further.
Combination Strategies for Resistance Management: The company is exploring combination therapies involving furmonertinib and other inhibitors like SHP2 to manage acquired resistance, a common hurdle in cancer treatment.
Leverage Innovative Platforms: ArriVent intends to work on antibody-drug conjugates (ADCs) via a research collaboration with Aarvik, aiming to identify lead candidates with improved activity and safety profiles.
Business Development for Pipeline Expansion: Their business strategy involves acquiring additional therapeutic candidates beyond furmonertinib, targeting solid tumors and other significant unmet medical needs in the oncology space.
Similar Approaches & Risks:
ArriVent's strategy is not unique; several biotech companies have pursued similar paths, especially with the recent focus on precision oncology. Examples include Blueprint Medicines, Loxo Oncology (acquired by Eli Lilly), and Turning Point Therapeutics.
Risks and pitfalls of the approach can include:
Clinical Risks: There is no guarantee that the drugs in their pipeline will prove to be safe and effective in later-stage clinical trials, or that they will receive regulatory approval.
Regulatory Risks: Even with a clear regulatory strategy, the rapidly changing landscape of drug approvals can introduce unexpected challenges or delays.
In-licensing Limitations: Depending on the terms of in-licensing agreements, ArriVent may face limitations in how they can develop and commercialize the drugs.
Market Competition: Given the intense competition in oncology, there is always a risk that competitors may develop more effective treatments or achieve regulatory approval first.
Intellectual Property Risks: They will need to navigate complex intellectual property landscapes and may face potential litigation or challenges to their patent claims.
Financial Risks: The development of novel therapeutics requires substantial investment, and there's always the risk that the company may not secure sufficient funding to support their programs through to commercialization.
In summary, ArriVent BioPharma's scientific strategy revolves around leveraging their expertise in oncology drug development and business development to build a competitive pipeline, mainly focusing on therapeutics originating from China's burgeoning pharmaceutical market. Their approach resembles other oncology-focused entities within the industry but comes with a set of risks inherent to the high-stakes world of drug development and commercialization.